
Content
Quality Risk Management (QRM) is no longer just a tool, but a cornerstone of the entire Pharmaceutical Quality System (PQS).
I've highlighted the changes for you in this carousel. Give it a swipe! ➡️
I wholeheartedly applaud the growing emphasis on QRM throughout the chapter. It now more explicitly calls for a proactive, risk-based approach to quality.
This is the very essence of what we do at CAPA Consulting with our QA Optimization services. We leverage QRM as a powerful, systematic process to make informed, risk-based decisions that protect patients and improve processes and business. Seeing this philosophy being embedded deeper into EU GMP is fantastic.
The future of quality is clear: it will be increasingly risk-based. QRM is the key to building more resilient, efficient, and reliable systems, and its importance will only continue to grow. Rightfully so.
What are your thoughts on the evolution of QRM in the industry?
📌 Note: Chapter 1 is currently in draft (stakeholders consultation) until 3rd of December. Nonetheless, it is highly unlikely that the content will undergo major changes from here.

EU GMP Chapter 1 PQS updated draft: In a nutshell
An overview of the new Chapter 1 draft on Pharmaceutical Quality System (PQS). Date: 08/09/2025. *Modifications in red. Date of creation: 08/09/2025

1. Principle
The Marketing Authorisation Holder (MAH) must manufacture medicinal products, ensuring that they are: Fit for their intended use; Comply with the requirements of the Marketing Authorisation (or Clinical Trial Authorisation); Do not place patients at risk due to inadequate safety, quality or efficacy. Attaining this so-called “quality objective” is the responsibility of senior management and requires commitment by staff at all levels. The documented and continuously monitored Pharmaceutical Quality System (PQS) is there to achieve this objective reliably. A proactive approach to QRM is of strategic importance in: achieving an effective PQS; achieving continuous improvement; enabling informed and timely decisions throughout the product lifecycle. The basic concepts of Quality Management, GMP and QRM are inter-related. The use of risk-based drug shortage prevention and mitigation activities with respect to product quality/manufacturing risks should be considered.

2. PQS
Quality Management is the sum total of the organised arrangements made with the objective of ensuring that medicinal products are of the quality required for their intended use. It covers all matters which collectively influence the quality of a product. GMP applies throughout the product lifecycle (from manufacturing of IMPs to tech transfer, to commercial manufacturing and to product discontinuation). A PQS should be commensurate the size and complexity of the activities. The design should incorporate Risk Management principles. Consistent delivery of products with appropriate Quality Attributes by designing, planning, implementing, maintaining and continuously improving the PQS.

What should a PQS ensure? (1/2)
A PQS should ensure: Management of product and process knowledge throughout the lifecycle. Development of medicinal products in line with GMP requirements. Manufacturing and QC activities in line with GMP requirements. Managerial responsibilities are documented. The manufacture, supply and use of the correct starting and packaging materials. The appropriate selection and monitoring of suppliers. Any outsourced activities are appropriately managed. A state of control is established and maintained by developing and maintaining monitoring and control systems; QRM should be used in the design and validation/qualification of such systems. The results of product and process monitoring are taken into account in batch release, in the investigation of deviations, and, with a view to taking preventive action to avoid potential deviations occurring in the future. All IPCs, relevant checks/analyses during manufacturing and any relevant validations are carried out. Continuous improvement.

What should a PQS ensure? (2/2)
A PQS should ensure: An appropriate level of root cause analysis is applied during the investigation of deviations, product defects and other problems. Appropriate corrective actions and/or preventive actions (CAPAs) should be identified and taken in response to investigations. The effectiveness of such actions should be monitored and assessed, in line with Quality Risk Management principles. Medicinal products are not sold or supplied before a Qualified Person has certified each production batch. Medicinal products are stored, distributed and subsequently handled so that quality is maintained throughout their shelf life. There is a process for self-inspection and quality audits. QRM, together with knowledge management, is used to provide an early warning system that supports oversight and response to evolving quality/manufacturing risks from the company or its external partners, including potential product shortage issues. Prospective evaluation of planned changes and their approval prior to implementation. After implementation of any change, an evaluation is undertaken to confirm the quality objectives were achieved and that there was no unintended deleterious impact on product quality.

Senior Management
Senior management has the ultimate responsibility to ensure an effective PQS is in place, adequately resourced and that roles are defined. Senior management’s leadership and active participation in the PQS is essential. This leadership should ensure the support of staff at all levels within the organisation to the PQS. There should be periodic management review (with the involvement of senior management) of the operation of the PQS to identify opportunities for continuous improvement of products, processes and the system itself.

3. GMP (1/2)
Good Manufacturing Practice is that part of Quality Management which ensures that products are consistently produced and controlled to the quality standards. Basic requirements are: All manufacturing processes are defined, systematically reviewed, and shown to be capable of consistently manufacturing products of the required quality. Manufacturing processes and significant changes are validated. All necessary facilities are provided (qualified personnel, adequate premises/equipment/services/materials, approved procedures, suitable storage/transport). Instructions and procedures are clear and unambiguous. External product availability risks are adequately managed. Procedures are carried out correctly by trained operators. Records are made during manufacturing and distribution to demonstrate all steps were taken and product quantity/quality is as expected; records are retained in a clear, accessible form. Any significant deviations are fully recorded, investigated for root cause, and appropriate CAPA is implemented.

3. GMP (2/2)
The basic GMP requirements are: The distribution of the products minimises any risk to their quality and takes account of Good Distribution Practice. A system is available to recall any batch of product, from sale or supply. Complaints about products are examined, the causes of quality defects investigated and appropriate measures taken in respect of the defective products and to prevent reoccurrence.

4. QC
Quality Control is the part of GMP concerned with sampling and testing, so that materials are not released for use, nor products released for sale, until their quality has been judged satisfactory. Basic QC requirements include: Adequate facilities, trained personnel, and approved procedures for sampling and testing materials and monitoring environmental conditions. Test methods are validated. Records demonstrate all required procedures were carried out and materials are assessed against specification. Finished products contain active ingredients compliant with the Marketing Authorisation, are of the required purity, and are correctly contained and labelled. No batch is released prior to certification by a Qualified Person. Sufficient reference samples of starting materials and products are retained.

5. PQR
Periodic Quality Reviews of all authorised medicinal products are conducted to verify the consistency of the process and appropriateness of specifications, to highlight trends, and to identify product and process improvements. Such reviews should take into account previous reviews and include at least: A review of starting and packaging materials (especially supply chain traceability of active substances). A review of in-process controls and finished product results. A review of all batches that failed to meet specification and their investigation. A review of all significant deviations and the effectiveness of resultant CAPAs. A review of all changes made to the processes or analytical methods. A review of Marketing Authorisation variations submitted, granted, or refused. A review of the stability monitoring programme and any adverse trends. A review of all quality-related returns, complaints and recalls. A review of adequacy of any other corrective actions. A review of post-marketing commitments for new authorisations and variations. The qualification status of relevant equipment and utilities. A review of any contractual arrangements.

5. PQR - Data Usage
Trending data from the previous product quality review should be included where few batches were manufactured in a 12-month period to ensure a more extensive data set. It can also be useful when a larger number of batches is manufactured. If no batches were manufactured during the review period, the PQR should still be performed. This review should address: stability results, returns, complaints, recalls, relevant deviations, and regulatory background (variations, commitments). A review of the last PQR should also be conducted. Use available data meaningfully. No recent activity still requires a PQR.

5. PQR - Timeframes & Grouping
Review timeframes can be adjusted based upon manufacturing and campaign duration, with adequate justification, and criteria should be established in a procedure. The Marketing Authorisation Holder should evaluate the results of the review and assess whether CAPAs or any revalidation should be undertaken, with procedures for ongoing management. Quality reviews may be grouped if scientifically justified and it enhances the overall review, but the strategy should not impede detection of adverse trends for an individual product. Reviewing only a representative or worst-case product is not acceptable.

6. QRM
Quality risk management is a systematic process for the assessment, control, communication and review of risks to the quality of the medicinal product. Product quality should be assured based on appropriate risk-based decision making throughout the product lifecycle. Knowledge should be used to make informed decisions, trigger re-evaluations and stimulate continuous improvement. The evaluation of risk to quality is based on scientific knowledge, experience, and ultimately links to patient protection. Risk to quality includes situations where product availability may be impacted. Each site in the supply chain should manage any manufacturing/quality risks that could impact product availability. The level of effort, formality and documentation of the QRM process should be commensurate with the level of risk.

6. QRM - Process
The output/results of the quality risk management process should be reviewed to take into account new knowledge and experience, with a mechanism to review and monitor events. Subjectivity can directly impact the effectiveness of QRM activities and should be managed and minimised. An appropriate level of formality should be applied, allowing less formal means for lower risk issues, freeing up resources for higher risk issues that require more rigor and effort.

Need Support?
Contact Nicolas Van Leeckwyck (apr.), Qualified Person, Founder of CAPA consulting. Offering QP/RP/QA services and QA optimization. Phone: +32 477 31 49 80, Email: nicolas@capaconsulting.be, Website: https://capaconsulting.be. Quality is an investment, not a cost.
September is here.
As a QA’er and a parent, I proudly say: “My daughter returns to school to continue her Training Plan.”
In Quality Management, it is no different.
Every standard and regulation requires training. GMP, GDP, ISO13485, the MDR…
If you boil it down, they all state the same requirement:
Initial Training + Continuous Training
Why?
Training is what transforms any SOP into Operation. The best-written SOP is useless if it’s not applied.
✅ A strong training system includes:
- Training Matrix – mapping “ideal profile” vs. “current profile.”
- Training Plan – personal plan for every employee (especially useful during onboarding).
- Training Logs – chronological logs per individual (your inspector’s first stop, so keep them clean!).
- Training Records – details for each training: date, duration (track as a KPI), subject, materials, trainer and attendees.
Oh and… please do not forget the Verification of Effectiveness. Organizing a training without verifying its effectiveness is like running a fire drill without checking if anyone evacuated. It just doesn’t make sense.
👉 The Reality:
Training takes a lot of time and energy.
That’s why CAPA Consulting helps QA teams by taking training management off their shoulders.
Outsourcing means:
- ✔️ Trainings designed to deliver the right message in the right way to each target audience.
- ✔️ A training system that is always inspection-ready.
- ✔️ More time and focus for you to tend to other parts of your QMS.
👉 Curious: how do you make sure training actually sticks in your organization?

In Quality roles, you frequently handle personal data (complaints, employee info, vigilance data,…). I always knew GDPR applied but never fully understood its scope or practical implications. After taking the time to dive into the regulation, I’ve identified 2 key definitions and 4 general rules that provide a solid foundation for understanding GDPR from a QA perspective.
📘 2 Definitions You Should Know:
- Personal data = any information relating to an identified or identifiable natural person (name, location, IP address, telephone number,…)
- Processing = any operation which is performed on personal data (recording, storage, use, dissemination,…)
✅ 4 General Rules for QA Professionals:
Rule No. 1 📢:
If you collect personal data, you must provide clear information to the individual (see art. 13).
Rule No. 2 ⚙️:
If you process personal data, stick to the 7 GDPR principles (see art. 5):
- Lawfulness, Fairness & Transparency: Be clear, honest, and lawful.
- Purpose Limitation: Only use data for the reason you collected it.
- Data Minimization: Collect only what you need.
- Accuracy: Keep data correct and up to date.
- Storage Limitation: Don’t keep data longer than necessary.
- Integrity & Confidentiality: Keep data secure and protected.
- Accountability: Be able to show you're compliant with GDPR.
Rule No. 3 🧾:
Lawful processing of personal data requires one of these six legal bases:
- Consent: The individual has clearly agreed to the processing.
- Contract: Necessary to perform or prepare a contract.
- Legal Obligation: Required to meet a legal duty (pharmacovigilance etc).
- Vital Interests: Needed to protect someone’s life.
- Public Task: Carried out in the public interest or under official authority.
- Legitimate Interests: For a valid business reason and only if not in conflict with individual privacy rights.
❗ Important Note: If you’re dealing with data concerning health, there are higher standards (see art. 9).
Rule No. 4 👮:
If you process personal data as part of your core activities (clinical trial data for instance), get a DPO (Data Protection Officer) and conduct a Data Protection Impact Assessment.
🚨 What happens if I don’t play by the rules?
You risk being publicly fined. Among the fined Belgian entities I found a hospital (€200k), a medical lab (€20k), the Belgian Order of Pharmacists (€30k),…
👉 Have a look yourself:
https://www.enforcementtracker.com/

The CAPA system is the most beautiful system a QMS has to offer (granted, I might be a bit biased). It enables organizations to plan, execute, verify effectiveness, and document actions that prevent the (re-)occurrence of systemic issues. In other words: it’s THE critical driver of continuous improvement.
But… there’s a risk.
Death By CAPA is the phenomenon where, for a prolonged period of time, CAPAs pile up faster than they’re being closed. As the pile grows, usually so do the delays in closure… resulting in:
- Resource strain
- Bad metrics (which have a secondary effect during audits/inspections)
- Prolonged exposure to risk
- And (often overlooked but very real) impact on staff morale and engagement
Needless to say that these effects gradually ripple into the financial performance of your organization.
How do you know if your organization is at risk?
It’s not always black or white, but telltale signs include overdue CAPAs, repeated deadline extensions, and a growing sense of “never catching up.”
👉 If this sounds familiar, you’re not alone. It’s exactly the type of challenges we help you tackle at CAPA Consulting.
From backlog removal to process redesign, we bring you the results you need.
If your CAPA system is costing you more than it’s saving, let’s fix that.
👇 Link in the comments below, or send a DM.

Not strictly a QA topic today, but definitely a close cousin.
Nutrivigilance is often intertwined with the complaint handling process, yet still flies under the radar for many.
📌 What is Nutrivigilance?
It’s a vigilance system designed to rapidly identify undesirable effects linked to the consumption of certain foodstuffs.
Think pharmacovigilance for medicinal products or materiovigilance for medical devices, but applied to specific food products.
In my experience, Nutrivigilance is too often:
❌ Overlooked
❌ Seen as “less important”
❌ Pushed down the priority list
But here’s the reality:
✅ It became a legal obligation for many actors in the Belgian life sciences industry in December 2023.
In the slide deck below, you’ll find:
- What Nutrivigilance is
- Why it matters
- Which products are in scope
- Who’s involved
- How proper reporting works
💡 Last slide bonus: 7 practical tips for safe use of dietary supplements.
If you’re involved and unsure whether your Nutrivigilance process is compliant, or if you need guidance building/supporting one:
📩 Reach out here on LinkedIn or follow the link in the comments.

Nutrivigilance: In a nutshell
RD 03 December 2023 — An overview of nutrivigilance, its purpose, scope, and key stakeholders. Date: 11 August 2025.

What & Why?
Introduction to the concept of nutrivigilance, covering its purpose, legal basis, and the importance of monitoring undesirable effects related to nutrition-related products.

Products in Scope
This includes: - Food supplements - Foodstuff for specific groups - Foods for special medical purposes - Novel foods - Fortified foods

Stakeholders
Operators: Any company involved in the product chain from manufacturing to availability to the consumer. Examples: Manufacturers, Distributors, Retailers. Legally obligated to report all undesirable effects within 10 working days.
Healthcare Professionals: Doctors, Pharmacists, Dieticians, Nurses, Dentists, Speech Therapists, Physiotherapists, and others. Not legally obligated but expected to report undesirable effects when made aware.
Citizens: Anyone who has knowledge of any undesirable effect, even if not personally experienced. Not legally obligated but can report undesirable effects when made aware.
Organisations of Public Interest & Public Utility Companies: Legally obligated to report all undesirable effects at least once per month.

Reporting
Concomitant medication: Nutrivigilance works in collaboration with Pharmacovigilance.
Scope: “Any suspected side effect that occurs on Belgian territory.”
This includes: - Reporter - User - Product - Undesirable effect - Associated use
Relevant attachments should be included with the report to provide complete context and documentation.

Need Support?
This slide indicates where to seek help for reporting or understanding nutrivigilance processes. It includes all relevant contact details of CAPA Consulting.

A Few Tips for Safer Consumption
Dietary supplements are not medicinal products and are not intended to prevent or treat disease. They should not replace medical treatment. If you are ill, consult your doctor.
Be wary of miracle products and advertisements that try to convince you that a dietary supplement or other food product can cure or prevent illness.
Ask your doctor or healthcare professional for advice before taking supplements.
Follow the quantity, conditions of use, recommendations, and warnings given on the label.
Take care when combining dietary supplements with medicinal products or other food supplements. If in doubt, consult your doctor or pharmacist.
Avoid prolonged or repeated use of dietary supplements.
Avoid buying dietary supplements or other food products from abroad via the internet.
I often compare a QMS system to a sailing ship. The goal of the QA team? Keep it afloat and on course (even when the weather gets bad or there’s a few holes in the hull).
However, when the Competent Authorities start losing trust in your system, you are placed under Compliance Management. Once you’re under it… getting out requires a lot (!) of additional effort.
🔍 So... What is Compliance Management?
It's a formal escalation step taken by the CA when inspectors determine that routine inspections are no longer sufficient to ensure GMP/GDP compliance. In other words, your ship is now in a vortex.
Once this process is triggered, you’re looking at:
- Increased inspection frequency (to every few months)
- Periodic monitoring and reporting (progress reports, compliance metrics,...)
- Cautionary letters and Conditioned approvals
❗ I’m in Compliance Management… Now what?
Either you successfully return to a state of compliance within the predefined period.
Or you fail to implement the required improvements within the predefined period,
leading to Regulatory Action (suspension of license for X months, license revoked, publication of a statement of non-compliance,… the bad stuff).
📈 How do you get out of it?
👉 In short, you need to regain trust from the CA. We can help you with that by providing the required additional wind in your sails. CAPA Consulting has first-hand experience in dealing with compliance management.
As always, it's better to prevent than to cure.

A few weeks ago, I shared some initial thoughts on the upcoming Annex 22 and its impact on the GMP environment.
📊 I'm now pleased to share a dedicated slide deck that explores Annex 22 in more detail.
🧠 In addition, a training session will be organised in collaboration with UPIP-VAPI – Belgian Professional Association of Pharmacists working in the Life Science Industry in the coming months, covering:
- The new Annex 22 (AI in GMP)
- The updated Annex 11 (Computerised Systems)
- The revised Chapter 4 (Documentation)
👉 Stay tuned if you’d like to be notified when registration opens.
If you’re currently working with AI in a GMP context, or need support aligning your processes with Annex 22 (or any other GMP requirements), feel free to reach out. I’d be happy to assist.
📌 Note: The annex 22 is currently in draft (stakeholders consultation) until 7 October. Nonetheless, it is highly unlikely that the content will undergo major changes from here.
Let’s keep the conversation going. 💬

EU GMP New Annex 22 (draft): Artificial Intelligence Overview
This slide introduces Annex 22, focused on Artificial Intelligence within GMP. Date: 29 July 2025.
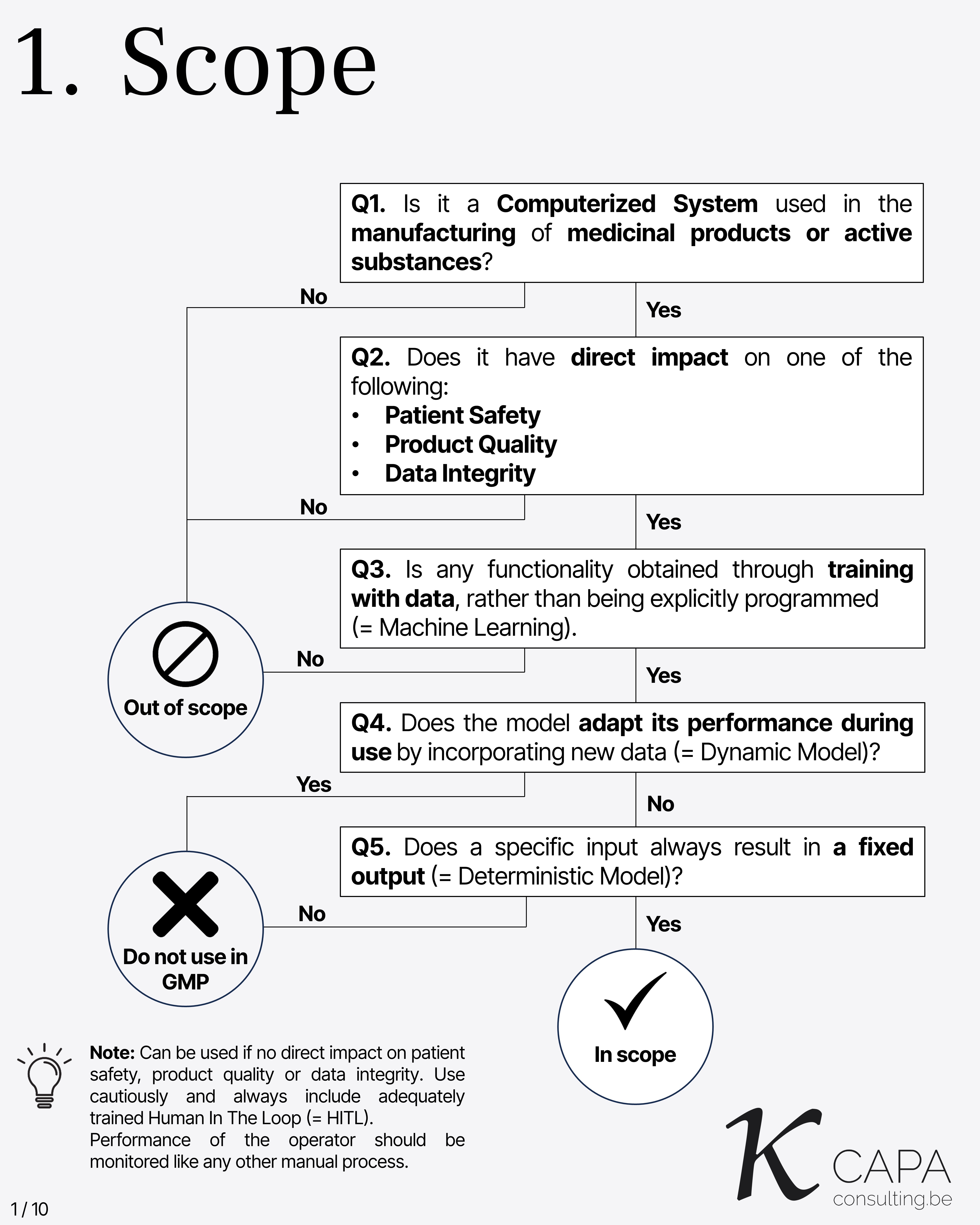
Scope
Key questions to determine if a computerized system is subject to Annex 22: 1. Is it used in manufacturing of medicinal products or active substances? 2. Does it directly impact patient safety, product quality, or data integrity? 3. Is functionality obtained through data training (Machine Learning)? 4. Does the model adapt over time with new data (Dynamic Model)? 5. Does a specific input always yield a fixed output (Deterministic Model)?
Note: If no direct impact, it may be used cautiously with a trained Human In The Loop (HITL), whose performance must be monitored.

Principle
The goal is understanding intended use and associated risks. Requirements include: - Close cooperation across all involved parties during algorithm selection, training, validation, testing, and operation. - Adequate qualifications and responsibilities. - Defined and appropriate access levels. - Availability and review of all documentation, even from third parties. - Quality Risk Management must assess risks to patient safety, product quality, and data integrity.

Intended Use
This slide defines the scope and purpose for which the AI model is implemented within GMP-regulated environments.

Acceptance Criteria
Test metrics (e.g., confusion matrix, accuracy, precision, F1-score) must be defined. Acceptance criteria must be clearly documented and assigned to the SME. If replacing an existing process, model performance must be at least as good as the original process.

Test Data
Test data must represent the full sample space, including all subgroups and variations. It should be large enough for statistically reliable metrics. Important considerations: - Pre-processing must be defined. - Any data exclusions must be documented. - AI-generated data is discouraged and must be justified if used. - Labels must be verified with high correctness.

Test Data Independence
Test data must be completely independent from training and validation data. Staff who accessed test data should not participate in training/validation. Access must be controlled, logged, and documented. No unauthorized copies of test data are allowed outside the designated repository.

Test Execution
The test must confirm model suitability for intended use and detect overfitting or underfitting. A formal test plan is required, including: - Intended use summary. - Pre-defined metrics and criteria. - Reference to test data. - Step-by-step test script. - Metric calculation methods. All deviations and failures must be documented and justified.
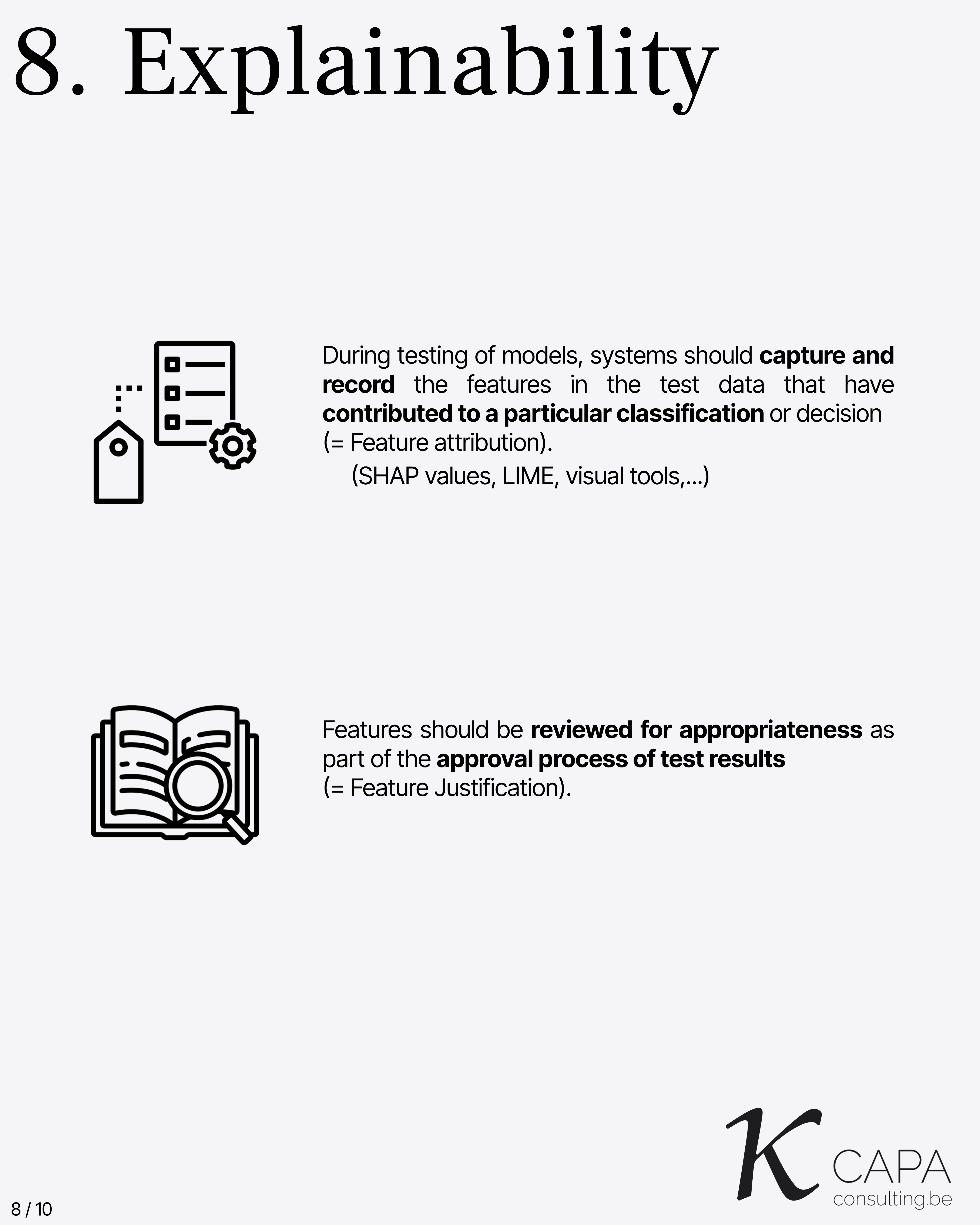
Explainability
During model testing, the features influencing decisions must be recorded (feature attribution). These features must be reviewed and justified (feature justification). Tools like SHAP values, LIME, or visual explanations may be used.

Confidence
This slide likely covers the importance of understanding and evaluating the confidence level of AI model outputs. Confidence metrics or scores should be available and interpreted appropriately to support reliable decision-making. Where confidence is low, human review or additional safeguards may be necessary.

Operations
Once a model has passed testing, it must be placed under configuration control before it is deployed in operational environments. This ensures protection against unauthorized changes.
Ongoing monitoring of the model’s performance is essential. It should include: - System performance monitoring to detect degradation or anomalies. - Input sample space monitoring to check if incoming data remains within the range the model was trained for. - Detection of data drift, where input distributions shift over time.

Need Support?
This closing slide invites viewers to seek support if needed. It implies the availability of resources or contacts to assist with implementation or interpretation of Annex 22 requirements related to Artificial Intelligence.
Deviations, Complaints, Out-Of-Specs, Reworks, Recalls, Breakdowns, Rejects,…
They all come with a hefty price tag 💸 if you really think about it.
At CAPA Consulting, we create new and optimize existing QMS systems based on
independent risk assessments.
The goal? To keep your operational cost of QA activities as low as possible without compromising compliance. ✅
📩 Want to hear more?

Yesterday, I attended the EIPG webinar on the revision of the Good Distribution Practice (GDP) guidelines. Some interesting insights were shared:
🔍 Key Takeaways:
- GDP will no longer be a guideline — it’s becoming an EU Regulation.
- 📌 Harmonisation across EU member states is the main objective.
- The EMA is responsible for drafting the text, based on input from the Inspectors Working Group.
👤 What does this mean for Responsible Persons (RPs)?
- For the first time, the role and responsibilities of the RP will be defined directly in EU legislation.
📜 New focus areas include:
- The use of AI
- Cybersecurity measures (!)
- Reducing audit fatigue through Joint Audits
📅 Expected Implementation Timeline: 2027 – 2028
👏 Special thanks to European Industrial Pharmacists Group for organizing this webinar which offered an exclusive look behind the scenes.
Key Details:
- 📅 Consultation Window: July 7 – October 7, 2025
- 🔍 Scope:
- Revised Annex 11 (Computerised Systems)
- Updated Chapter 4 (Documentation)
- Brand-new Annex 22 (AI in Pharma Manufacturing)
- 🏛️ Developed by: EMA & PIC/S to harmonize digital/GMP standards globally.
Highlights of Proposed Changes
🔬 NEW Annex 22: AI in GMP
- Scope: Only static, deterministic AI models permitted for critical applications (e.g., product release).
- Exclusions: Generative AI/LLMs banned from critical processes; "human-in-the-loop" required for non-critical uses.
- Critical Rules: Model explainability (e.g., SHAP/LIME), confidence scoring, and lifecycle monitoring.
🔒 Revised Annex 11: Computerised Systems
- Security: Mandates MFA, penetration testing, and network segmentation.
- Data Integrity: Audit trails now required (not risk-based).
- Quality Integration: Direct links to Pharmaceutical Quality Systems (PQS) for deviations/CAPA.
📄 Revised Chapter 4: Documentation
- ALCOA++ Principles: "Traceable + Enduring" standards for all data (text, images, audio).
- Hybrid Systems: Clear governance for electronic signatures and cloud-based solutions.
- Lifecycle Control: Risk-based retention/disposal protocols.
Why This Matters
These updates modernize EU GMP for the digital age — integrating AI governance, cybersecurity rigor, and end-to-end data integrity. Annex 22 sets a pioneering standard for trustworthy AI in pharma.
🔜 Coming Soon: I’ll break down the implications of each proposed change in more detail. Stay tuned!
(Less “Oops” — more “Aaaah!”)
Waiting for problems to hit wastes time, money, and can jeopardize patient safety. Instead, shift to a proactive quality mindset — where issues are prevented before they happen. It’s a game-changer for compliance and efficiency of your QMS.
🔍 Key Contrasts:
- Reactive: Occurrence → Detection → Correction
(High cost, Always late, Firefighting mode) - Proactive: Assess → Prevent → Review
(Low cost, Forward-thinking, Quality-driven culture)
✅ Why Proactive Wins:
- Cost Savings – Prevention slashes correction costs and waste
- Safety & Compliance – Fewer deviations and failures
- Regulatory Alignment – Most modern GxP guidelines support and encourage risk-based strategies
👉 Shift Your Mindset:
Go from “Are we compliant enough?” to “How do we prevent failure tomorrow?”
💬 Interested in going proactive?
Need tailored support? CAPA Consulting can help optimize your Quality System.
It was an incredible experience:
An international project, a top-tier facility, and the opportunity to work alongside two brilliant co-auditors (and now, new friends).
Audits aren’t just about compliance — they’re moments of cross-pollination.
I learned a lot.
I shared a lot.
And most rewarding of all: I witnessed teams thinking together to solve findings not by adding more procedures, but by improving the design of the existing system.
That's real quality culture in action. 🙌
Beyond the audit, I gained a deeper appreciation for the local culture — The hospitality, work ethic, and the commitment to continuous improvement truly stood out.
If you’re looking for support with EU GMP or GDP audits — whether in Europe or abroad — don’t hesitate to reach out. I’d be happy to support you.

it was a great reminder of how essential training is in maintaining a strong quality culture.
As outlined in the GDP guidelines:
"Personnel should receive initial and continuing training relevant to their role…. The responsible person should also maintain their competence in GDP through regular training."
It's a simple statement — but there’s a lot behind it.
📚 Training is not just a requirement — it's a backbone of quality.
That perfect SOP is useless if stakeholders don’t know what’s in it, or worse, don’t follow it. That’s why training should never be treated as a checkbox exercise.
What resonated most? The real-world case studies. Let’s be honest: reality loves throwing curveballs ⚾️, and applying GDP principles often stretches beyond the written guidelines.
👏 Special thanks to Frank Peeters, Ludwig Everaert and Rizovsky Mounir for organising such a valuable event!
🔍 Do you struggle with training in your organisation?
Whether it's engagement, consistency, or real-world application — let’s talk. I’d love to hear where your challenges lie and explore how we can help.
P.S. Are you active as RP? Don’t forget to attend a GDP refresher training frequently — it’s a detail that inspectors always check during GDP inspections.

Got a question?
Get in touch via the link below.